One of the major causes of chronic kidney disease (CKD) is acute kidney injury, damage that can occur during bypass surgery, as a result of septic infection, after exposure to toxins, as a complication of diabetes, and even following short bouts of dehydration.
“Rather than maladaptive repair, there seems to be a distinct pathway of maladaptive de-differentiation that sustains the chronic kidney injury.”
Hoping to prevent that progression, cell biologist Craig Brooks, Ph.D., an assistant professor in the Division of Nephrology and Hypertension at Vanderbilt University Medical Center, is investigating a novel signaling pathway that may ultimately serve as a therapeutic target.
Brooks, who studies the molecular mechanisms that underlie poor self-repair by injured kidneys, and his team reported these latest findings in The Journal of Clinical Investigation.
“Rather than maladaptive repair, there seems to be a distinct pathway of maladaptive de-differentiation that sustains the chronic kidney injury,” Brooks said.
When Repair Goes Awry
Normally, the kidney has a remarkable capacity for healing following acute kidney injury, Brooks explained.
But an exception occurs when kidney epithelial cells lag in their anticipated recovery and drive fibrosis of the surrounding tissue, which in turn can promote CKD. This phenomenon, known for decades, has been called “maladaptive repair.”
“Many studies have sought to identify these maladaptively repaired cells and the processes that lead them to become pro-fibrotic,” Brooks said.
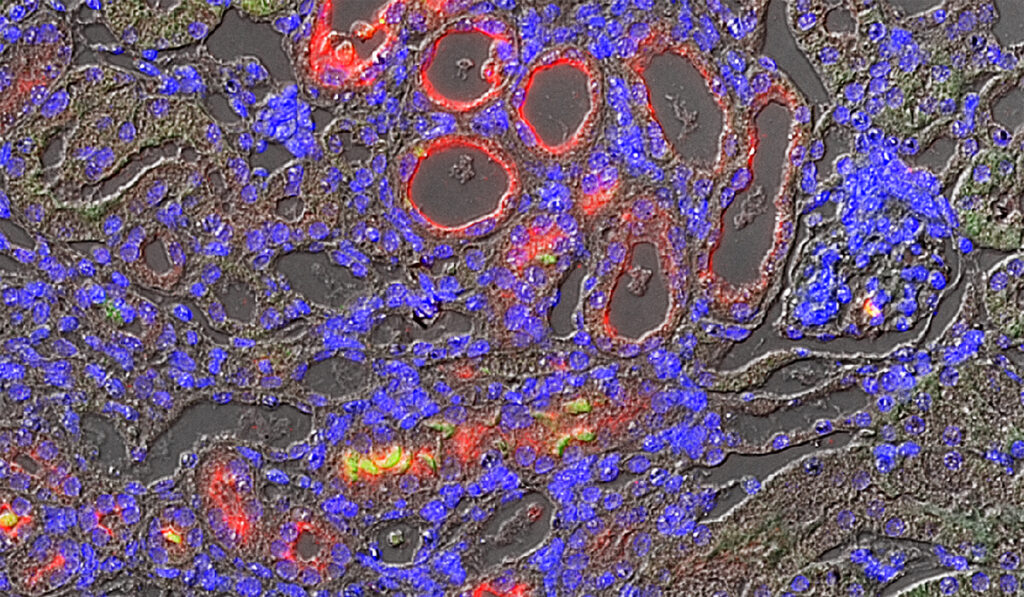
Brooks and his colleagues have used animal models to compare injured kidneys that progress to CKD against those that do not. In previous studies, they linked CKD to a particular phase of the kidney cell division cycle, where a large number of cells were found to be arrested. These cases were tied to development of CKD, with the researchers implicating the cell cycle protein cyclin G1.
Sustained Injury
In their current study, the researchers demonstrated that cyclin G1 is highly expressed in kidney tissue from patients with CKD and that it promotes fibrosis in animal models.
They also found, however, that cell cycle arrest is not critical to cyclin G1’s role in fibrosis and CKD. Rather, they showed that cyclin G1 acts through another protein called cyclin-dependent kinase 5 (CDK5) to promote kidney cell de-differentiation (a less-mature state), which drives fibrosis and CKD.
“It doesn’t appear to be a failure to repair; there’s a different process going on.”
De-differentiation is also required for kidney cells to repair after acute injury: Cells revert to an earlier developmental state so that they can divide and replace injured cells.
The researchers found that cyclin G1 and CDK5 do not regulate dedifferentiation associated with acute injury repair. Instead, associated signaling induces a prolonged state of de-differentiation that contributes to chronic disease.
“It doesn’t appear to be a failure to repair; there’s a different process going on,” Brooks said.
Targeting Differentiation
The findings suggest that therapeutics targeting the cyclin G1/CDK5 signaling pathway could be a novel approach to preventing CKD.
Brooks is also interested in testing cancer therapeutics that promote differentiation as a therapeutic strategy to prevent chronic disease in the injured kidney. Some cancer cells dedifferentiate to permit uncontrolled cell division, he said.
“The average survival on dialysis is only three to five years; better treatment options are needed to prevent chronic kidney disease progression,” Brooks said.