Abnormalities in fetal heart rate and rhythm complicate a small minority of pregnancies, but they can lead to fetal distress, prematurity and even stillbirth.
Many of these complications are related to long QT syndrome (LQTS), a heritable cardiac conductivity disorder that affects around 1 in 2,500 people and presents in 1 in 2,000 fetuses.
In 2020, pediatric and fetal cardiologist Stacy Killen, M.D., M.S.C.I., of Monroe Carell Jr. Children’s Hospital at Vanderbilt, participated in a groundbreaking multicenter, international study that illuminated the significantly heightened risk of stillbirth faced by mothers who have LQTS.
She is now on a quest to alert obstetricians to this fetal LQTS, the significance a low fetal heart rate for gestational age, and the need for definitive testing.
“Many times, we can identify a baby who might not survive to delivery, but through careful monitoring and transplacental therapy, achieve a normal-term delivery and an excellent quality of life after birth,” Killen said.
She recommends early identification and tracking of pregnancies complicated by fetal arrhythmias, followed by comprehensive risk-assessment and a customized a care plan, with informed decision-making throughout the process.
“For some fetal arrhythmias successfully treated in utero, the baby will have a normal heart rate and rhythm postnatally, without need for further cardiac intervention,” Killen said.
Hazards of Channelopathy
Congenital LQTS is a rhythm abnormality caused by mutations in genes encoding proteins in the sodium, potassium or calcium channels, which leads to their malfunction. In utero, LQTS can precipitate bradycardia, atrioventricular block, torsades de pointes, hydrops, and fetal death.
“Many times, we can identify a baby who might not survive to delivery, but through careful monitoring and transplacental therapy, achieves a normal-term delivery and an excellent quality of life after birth.”
Despite some de novo cases, congenital LQTS is primarily autosomal dominant, with the child of a carrier at a 50-a percent risk of inheriting the mutation, though the condition does not always manifest. Sometimes, recognizing LQTS in a fetus may lead to identification of a previously undiagnosed parent or sibling with the mutation.
In pregnant women with untreated LQTS, the condition presents potential hazards for the mother and the fetus, regardless of whether the condition has been passed on.
A Case Study
Killen and her mentor from the Medical College of Wisconsin, Janette Strasburger, M.D., recently diagnosed LQTS in an otherwise healthy fetus referred by Vanderbilt maternal-fetal medicine expert Soha Patel, M.D. Patel was the corresponding author for a report on the case published in JACC in February 2024.
While prenatal screening would not necessarily have flagged a LQTS problem, Patel incidentally noted low fetal heart rates – consistently below the third percentile for gestational age – in this otherwise unremarkable pregnancy. While the measurement didn’t reach the cut-off for diagnosing bradycardia (110-120 bpm), Patel became concerned and referred the case to Vanderbilt’s fetal cardiology team.
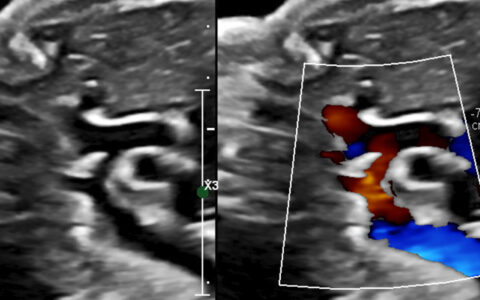
An echocardiogram uncovered no cardiac structural abnormalities but did find a prolonged left-ventricular isovolumic relaxation time in association with the previously observed low fetal heart rate. A series of two fetal magneto-cardiograms supported a LQTS diagnosis. At 31-weeks’ gestation, five-second fetal rhythm strips demonstrated QT prolongation and late peaking T waves.
After birth, genetic testing confirmed a variant in KCNQ1 consistent with the infant’s LQT1 phenotype. Parental testing identified that same variant in the mother, who did not have a known history of LQTS or a prolonged QTc on ECG. The mother’s twin sister was also alerted for testing.
Propranolol was initiated after birth, and both mother and baby continue on this therapy.
Early Detection, Treatment
In Patel’s patient, the heart rate was persistently low enough over time to raise an alarm. However, Killen says, fetal heart-rate ranges that fall short of the typical obstetrical definition of bradycardia may also be suspect.
New guidelines are evolving, with input from Vanderbilt’s Prince Kannankeril, M.D., M.S.C.I., who recently helped author the American Heart Association’s scientific statement on treatment of arrhythmias in fetuses and newborns, including guidance on screening for LQTS. He and Killen are collaborating on more research through a LQTS consortium to better understand how LQTS affects maternal and fetal health.
Further, Killen is participating in multicenter studies on the diagnosis and management of other fetal arrhythmias, including supraventricular tachycardia and anti-Ro/SSA-mediated heart block.